

-
Comparison of density functionals for energy and structural differences between the high- [5T2g: (t2g)4(eg)2] and low- [1A1g: (t2g)6(eg)0] spin states of the hexaquoferrous cation [Fe(H2O)6]2+
A. Fouqueau, S. Mer, M.E. Casida, L.M. Lawson Daku, A. Hauser, T. Mineva and F. Neese
Journal of Chemical Physics, 120 (20) (2004), p9473-9486


DOI:10.1063/1.1710046 | unige:3615 | Abstract | Article HTML | Article PDF | Article PS (gzipped)
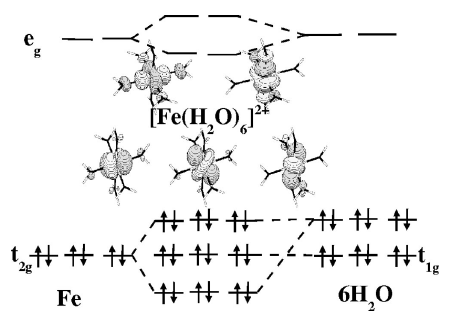
A comparison of density functionals is made for the calculation of energy and geometry differences for the high- [5T2g:â(t2g)4(eg)2] and low- [1A1g:â(t2g)6(eg)0] spin states of the hexaquoferrous cation [Fe(H2O)6]2+. Since very little experimental results are available (except for crystal structures involving the cation in its high-spin state), the primary comparison is with our own complete active-space self-consistent field (CASSCF), second-order perturbation theory-corrected complete active-space self-consistent field (CASPT2), and spectroscopy-oriented configuration interaction (SORCI) calculations. We find that generalized gradient approximations (GGAs) and the B3LYP hybrid functional provide geometries in good agreement with experiment and with our CASSCF calculations provided sufficiently extended basis sets are used (i.e., polarization functions on the iron and polarization and diffuse functions on the water molecules). In contrast, CASPT2 calculations of the low-spinâhigh-spin energy difference ÎELH = ELSâEHS appear to be significantly overestimated due to basis set limitations in the sense that the energy difference of the atomic asymptotes (5Dâ1I excitation of Fe2+) are overestimated by about 3000 cmâ1. An empirical shift of the molecular ÎELH based upon atomic calculations provides a best estimate of 12â000â13â000 cmâ1. Our unshifted SORCI result is 13â300 cmâ1, consistent with previous comparisons between SORCI and experimental excitation energies which suggest that no such empirical shift is needed in conjunction with this method. In contrast, after estimation of incomplete basis set effects, GGAs with one exception underestimate this value by 3000â4000 cmâ1 while the B3LYP functional underestimates it by only about 1000 cmâ1. The exception is the GGA functional RPBE which appears to perform as well as or better than the B3LYP functional for the properties studied here. In order to obtain a best estimate of the molecular ÎELH within the context of density functional theory (DFT) calculations we have also performed atomic excitation energy calculations using the multiplet sum method. These atomic DFT calculations suggest that no empirical correction is needed for the DFT calculations.